


RESOLUTION-1 (KDAF)
Phase 2b, Double-Blind, Placebo-Controlled Study to Evaluate Peresolimabin Adult Participants with Moderately-to-Severely Active Rheumatoid Arthritis
WEBSITE LINK for patient
QUICK BITS:
DIAGNOSIS: Rheumatoid arthritis (NO other inflammatory conditions/CTDs // fibromyalgia ok if NOT active)
Biologic Naïve: Not required. Does not need to be on cDMARDs during the study.
Failed Medications —> AT LEAST ONE OF THESE:
azathioprine
methotrexate
hydroxychloroquine
leflunomide
sulfasalazine
biologic disease-modifying antirheumatic drugs (bDMARDs)
targeted synthetic disease-modifying antirheumatic drugs (tsDMARDs)
Failed NO MORE THAN 3 bDMARDs/tsDMARDs
Serologies: +RF or CCP OR previous radiographs that show images of the hands or feet documenting bony erosions consistent with RA.
Labs:
CRP: >1.2 times ULN
GFR >= 60
Swollen & Tender Joints:
≥6 swollen joints based on 66 joint count, and ≥6 tender joints based on 68 joint count. Notes: The distal interphalangeal joints should be evaluated but not included in the total count to determine eligibility.
Active synovitis in ≥1 joint in hands or wrists at screening by MRI.
Washout Periods:
*** If the previous study intervention has a long half-life, 3 months or 5 half-lives, whichever is longer, should have passed prior to screening visit.
28 days prior to MRI:

8 weeks (or 12 months for B cell depleting therapy) prior to MRI:

NSAIDs: Stable dose for at least 7 days before planned randomization. Increases in dose or introduction of new NSAIDs are not allowed during the study.
Vaccinations: Up to date vaccinations per regional and national guidelines, specifically influenza, pneumonia, and zoster. Administered ≥30 days before randomization
Duration: 48 weeks
Open Extension: Yes
Placebo arm: Yes, cross over at 12 week
Rescue therapy: Yes at week 12 (under “Rescue Medicine” section of protocol)
Study timeline: Screening period // 12-week double-blind, placebo-controlled treatment period // 48-week double-blind treatment period, and // post-treatment follow-up
Joint assessor: BLINDED! (has to be medical professional)
STUDY PROTOCOL can be found here
STUDY PRODUCT:
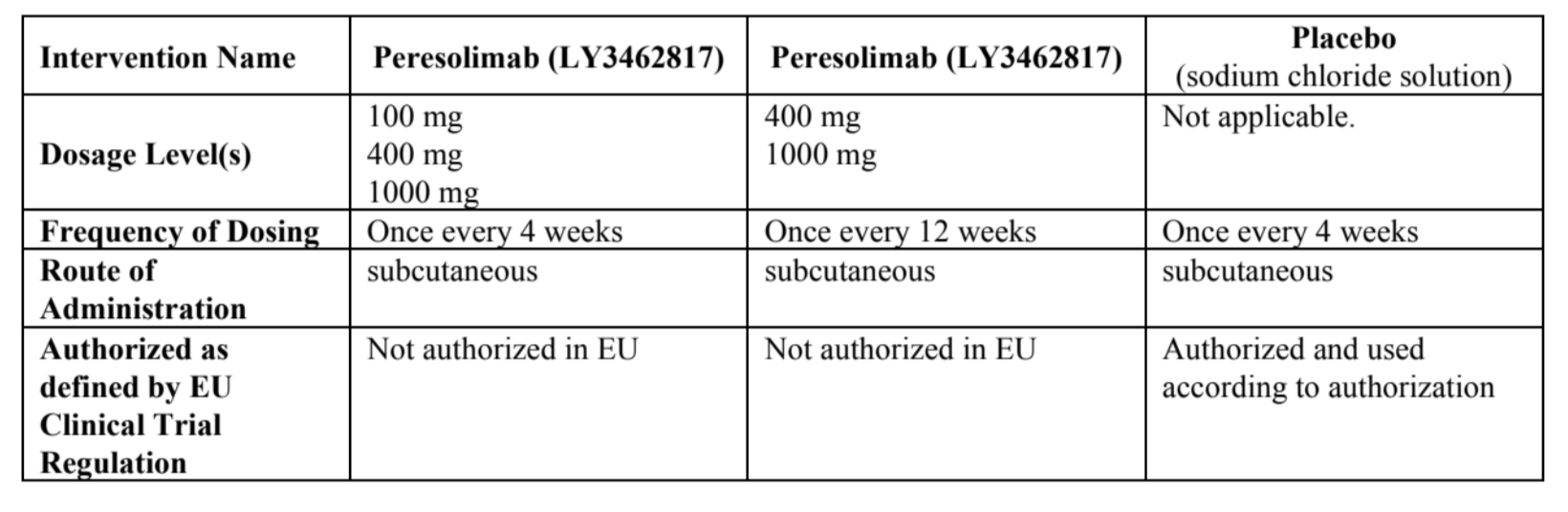
Peresolimab (LY3462817) in 100 mg, 400 mg, and 1000 mg Q4W vs Q12W
SUBQ (done ONSITE)
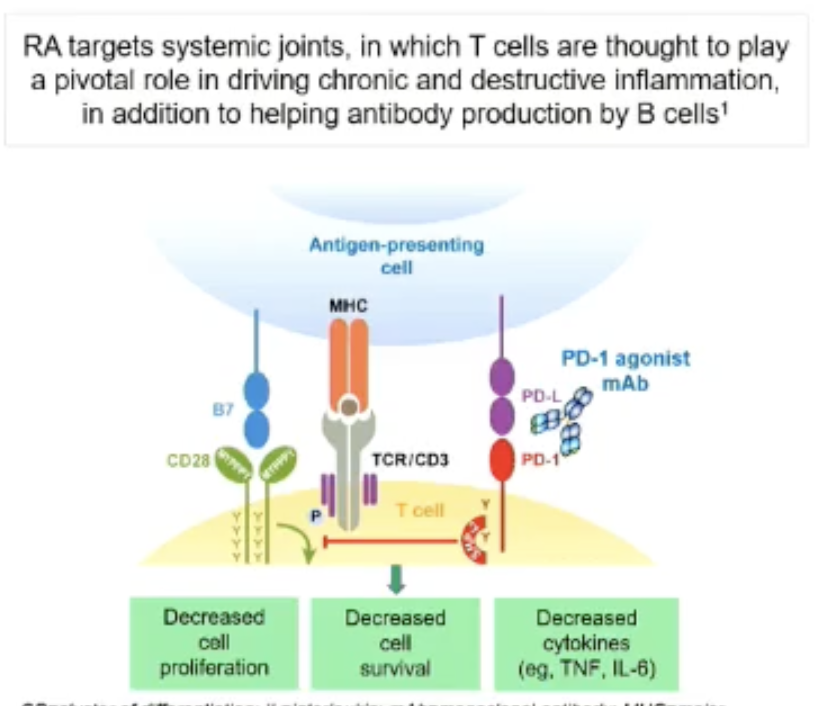
MOA: A humanized immunoglobulin G1 monoclonal antibody that stimulates human programmed cell death protein 1 (PD-1). The researchers hypothesized that the binding of peresolimab to PD-1 could stimulate physiological immune inhibitory pathways to restore immune homeostasis. (article here)
Most reported AE was headaches
TREATMENT GROUP IN A 1:2:2:1:1 RATIO:

{NOTE: in the diagram above, 1 mg should be 100 mg, 4 mg should be 400 mg, 10 mg should be 1000 mg}
PLACEBO: At Week 12, participants receiving placebo Q4W will begin receiving either 400 mg or 1000 mg peresolimab Q4W. Treatment assignment will be blinded and the participants will continue their assigned peresolimab dose and dosing frequency until the end of the study.
100 mg GROUPS: Continue their assigned dose and dosing frequency throughout the duration of the treatment period. No changes will occur.
400 mg or 1000 mg GROUPS: Continue their assigned dose and dosing frequency until Week 24. At Week 24, participants may be switched to Q12W dosing following assessment of CDAI scores, unless they received rescue medication prior to Week 24.
DOSAGE & ADMINISTRATION:
STUDY DURATION: up to 72 weeks
PRIMARY OBJECTIVE: Demonstrate that peresolimab is superior to placebo in achieving ACR20 at week 12.
SECONDARY OBJECTIVE: Secondary To evaluate the effect of peresolimab compared to placebo for measures of disease activity
BLINDING: Double-blind study. Blinding will be maintained throughout the conduct of the study
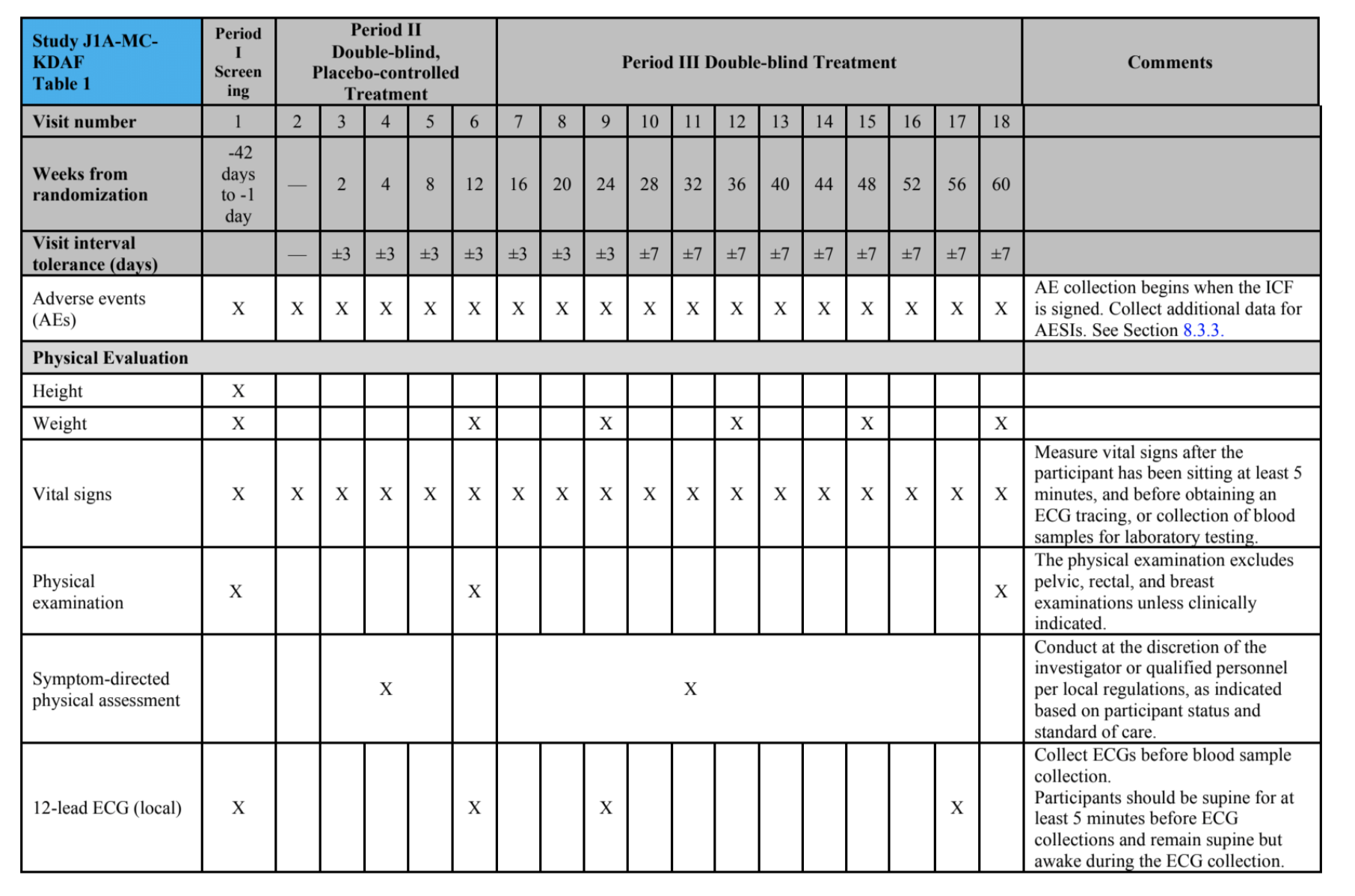
STUDY SCHEDULE:
📌 INCLUSION CRITERIA
1. Are ≥18 years of age at the time of signing the informed consent.
2. Have a diagnosis of adult onset RA for at least 3 months prior to screening, as defined by the 2010 ACR/European League Against Rheumatism.
3. Have moderately-to-severely active RA, at screening and baseline, defined by the presence of:
≥6 swollen joints based on 66 joint count, and ≥6 tender joints based on 68 joint count. Notes: The distal interphalangeal joints should be evaluated but not included in the total count to determine eligibility.
If a participant has received corticosteroid treatment per Criterion 27, the treated joint should be excluded from the joint count.
4. Positive RF or CCP OR previous radiographs documenting bony erosions in hands or feet consistent with RA.
5. CRP >1.2 times ULN per the central laboratory at screening.
6. Have active synovitis in ≥1 joint in hands or wrists at screening as demonstrated by an MRI.
7. Have up to date vaccinations per regional and national guidelines, specifically influenza, pneumonia, and zoster. • Live vaccines should be administered >3 months before randomization. • All vaccinations should be administered ≥30 days before randomization.
8. Have clinical laboratory test results within normal reference range or results with acceptable deviations that are judged as not clinically significant by the investigator at screening.
9. Have had an inadequate response to, or loss of response or intolerance to at least 1 csDMARD, bDMARD, or tsDMARD treatment (see list above // see Section 6.8 for allowed treatments during the study if the dose is stable for ≥4 weeks prior to the screening MRI)
10. Male or female Women of childbearing potential (WOCBP) and women not of childbearing potential (WNOCBP) may participate in this study. Males who agree to use highly effective methods of contraception may participate in this study. See Section 10.4 for contraception requirements
11. Are capable of giving signed informed consent
📌 EXCLUSION CRITERIA
12. Have Class IV RA according to ACR revised response criteria.
13. Have an abnormality in the 12-lead ECG that, in the opinion of the investigator, increases the risks associated with participating in the study.
14. Have presence of 1 or more significant concurrent medical conditions per investigator judgment, including but not limited to poorly controlled diabetes or hypertension, chronic kidney disease stage IIIb, IV, or V, symptomatic heart failure according to New York Heart Association Class II, III, or IV, myocardial infarction, unstable angina pectoris, stroke or transient ischemic attack, within the past 12 months before randomization, severe chronic pulmonary disease, for example, requiring oxygen therapy, major chronic inflammatory disease or connective tissue disease other than RA, including but not limited to, o systemic lupus erythematosus, psoriatic arthritis, axial spondyloarthritis including ankylosing spondylitis and nonradiographic axial spondyloarthritis, reactive arthritis, gout, scleroderma, polymyositis, dermatomyositis, active fibromyalgia, or multiple sclerosis.
15. Have a history of chronic alcohol abuse, IV drug abuse, or illicit drug abuse within 1 year before screening. Note: Marijuana use is prohibited during participation in this study, regardless of local laws or if used for medical purposes. Cannabidiol products may be used during the study if they are derived exclusively from hemp. Participants who use hemp-based cannabidiol products must be on a stable dose for at least 10 days prior to randomization, and participants must remain on that stable dose during the study.
16. Have a C-SSRS ideation within 1 month prior to screening or any suicidal behavior within 3 months prior to screening and either ideation or suicidal behavior during screening prior to Visit 2.
17. Have a diagnosis or history of malignant disease within 5 years prior to baseline, with the exceptions of basal cell or squamous epithelial carcinomas of the skin that have been resected with no evidence of metastatic disease for 3 years, or cervical carcinoma in situ, with no evidence of recurrence within the 5 years prior to baseline.
18. Have presence of confirmed cervical dysplasia.
19. Have estimated glomerular filtration rate from serum creatinine using the Modification of Diet in Renal Disease method of <60 mL/min.
20. Have had any surgical procedure, within 12 weeks prior to screening, or any planned surgical procedure scheduled to occur during the study, with the exception of minor surgery requiring local or no anesthesia and without any complications or sequelae.
21. Have had any of the following types of infections within 3 months of screening or develops any of these infections before the randomization visit.
Serious - requires hospitalization or IV or equivalent oral antibiotic treatment.
Opportunistic - as defined in Section 10.7.
Chronic - duration of symptoms, signs, or treatment of 6 weeks or longer.
Recurring - including, but not limited to, herpes simplex, herpes zoster, recurring cellulitis, or chronic osteomyelitis. Note: Herpes zoster is considered active and ongoing until all vesicles are dry and crusted over
Exception: If the investigator determines that a participant with recurrent nonserious infections such as cellulitis and uncomplicated orolabial or genital herpes, is not at an increased risk of complications.
22. Have any of these infections: HIV infection, current infection with HBV, for example, positive for hepatitis B surface antigen (HBsAg) or polymerase chain reaction (PCR) positive for HBV DNA, current infection with HCV, for example, positive for HCV RNA, or active TB.
23. Have or had LTBI that has not been treated with a complete course of appropriate therapy as defined by the WHO or the United States CDC, unless such treatment is underway, as per Section 8.2.8.
24. Have a current or recent acute active infection, or fever of 100.5°F (38°C) or above, at screening or baseline. For at least 30 days prior to screening, participants must have no symptoms or signs of confirmed or suspected infection and must have completed any appropriate anti-infective treatment.
25. Are women who are currently pregnant or breastfeeding, or who intend to become pregnant or to breastfeed at any time during the study or within 20 weeks after receiving the last dose of study intervention.
26. Have failed more than 3 advanced therapies, which includes bDMARDs and tsDMARDs.
27 and 28. Are currently receiving or have received any of these therapies prior to the screening MRI. Therapy (see above under WASHOUT PERIOD)
29. Have received live vaccine(s), including live attenuated vaccines, within 4 weeks prior to screening or intend to receive during the study and is within 5 half-lives after the last dose of intervention. Exceptions: Non-live or inactivated vaccine if received at least 2 weeks before baseline or after the last study visit Inactivated influenza and pneumococcal vaccines, and SARS-CoV-2 vaccines authorized by local regulatory bodies.
30. Have received a Bacillus Calmette-Guérin (BCG) vaccination or BCG treatment within 12 months of screening. Prior/concurrent clinical study experience
31. Were previously enrolled in a clinical study investigating peresolimab (LY3462817) or any other molecule targeting PD-1 for the treatment of auto-immune or autoinflammatory conditions.
32. Are currently enrolled in any other clinical study involving a study intervention or any other type of medical research judged not to be scientifically or medically compatible with this study.
33. Have participated, within the last 30 days, in a clinical study involving study intervention. If the previous study intervention has a long half-life, 3 months or 5 half-lives, whichever is longer, should have passed prior to screening.
34. Have previously completed or withdrawn from this study. Diagnostic assessments Other exclusions
35. Have contraindications to MRI: for example, claustrophobia, pacemakers, aneurysm clips, and intraocular metallic fragments, IV gadolinium diethylenetriamine penta-acetic acid, including but not limited to moderate or severe renal insufficiency or allergic reaction to gadoliniumcontaining contrast media.
36. Have donated more than a single unit of blood within 4 weeks before randomization or plan on donating blood during the study.
37. Are investigator site personnel directly affiliated with this study and/or their immediate families. Immediate family is defined as a spouse, parent, child, or sibling, whether biological or legally adopted.
38. Are Lilly employees or employees of third-party organizations involved with the study that require exclusion of their employees.
39. Are not willing to receive SC injections.
40. Are unsuitable for inclusion in the study, in the opinion of the investigator or sponsor, for any reason that may compromise the participant’s safety or confound data interpretation